Vardict Germline Variant Caller¶
vardictGermlineVariantCaller · 2 contributors · 1 version
No documentation was provided: contribute one
Quickstart¶
from janis_bioinformatics.tools.variantcallers.vardictgermline_variants import VardictGermlineVariantCaller wf = WorkflowBuilder("myworkflow") wf.step( "vardictgermlinevariantcaller_step", VardictGermlineVariantCaller( bam=None, intervals=None, sample_name=None, header_lines=None, reference=None, ) ) wf.output("variants", source=vardictgermlinevariantcaller_step.variants) wf.output("out", source=vardictgermlinevariantcaller_step.out)
OR
- Install Janis
- Ensure Janis is configured to work with Docker or Singularity.
- Ensure all reference files are available:
Note
More information about these inputs are available below.
- Generate user input files for vardictGermlineVariantCaller:
# user inputs
janis inputs vardictGermlineVariantCaller > inputs.yaml
inputs.yaml
bam: bam.bam
header_lines: header_lines
intervals: intervals.bed
reference: reference.fasta
sample_name: <value>
- Run vardictGermlineVariantCaller with:
janis run [...run options] \
--inputs inputs.yaml \
vardictGermlineVariantCaller
Information¶
URL: No URL to the documentation was provided
| ID: | vardictGermlineVariantCaller |
|---|---|
| URL: | No URL to the documentation was provided |
| Versions: | v0.1.1 |
| Authors: | Michael Franklin, Jiaan Yu |
| Citations: | |
| Created: | 2019-03-28 |
| Updated: | 2020-07-14 |
Outputs¶
| name | type | documentation |
|---|---|---|
| variants | Gzipped<File> | |
| out | VCF |
Workflow¶
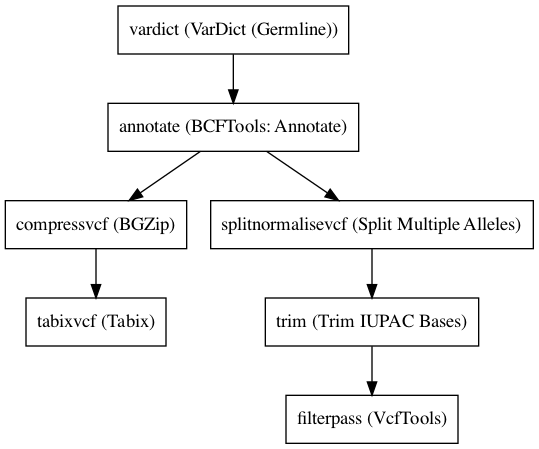
Embedded Tools¶
| VarDict (Germline) | vardict_germline/1.6.0 |
| BCFTools: Annotate | bcftoolsAnnotate/v1.5 |
| BGZip | bgzip/1.2.1 |
| Tabix | tabix/1.2.1 |
| Split Multiple Alleles | SplitMultiAllele/v0.5772 |
| Trim IUPAC Bases | trimIUPAC/0.0.5 |
| VcfTools | VcfTools/0.1.16 |
Additional configuration (inputs)¶
| name | type | documentation |
|---|---|---|
| bam | IndexedBam | |
| intervals | bed | |
| sample_name | String | |
| header_lines | File | |
| reference | FastaWithIndexes | |
| allele_freq_threshold | Optional<Float> | |
| vardict_chromNamesAreNumbers | Optional<Boolean> | Indicate the chromosome names are just numbers, such as 1, 2, not chr1, chr2 |
| vardict_vcfFormat | Optional<Boolean> | VCF format output |
| vardict_chromColumn | Optional<Integer> | The column for chromosome |
| vardict_regStartCol | Optional<Integer> | The column for region start, e.g. gene start |
| vardict_geneEndCol | Optional<Integer> | The column for region end, e.g. gene end |
| compressvcf_stdout | Optional<Boolean> | c: Write to standard output, keep original files unchanged. |
| filterpass_removeFileteredAll | Optional<Boolean> | Removes all sites with a FILTER flag other than PASS. |
| filterpass_recode | Optional<Boolean> | |
| filterpass_recodeINFOAll | Optional<Boolean> | These options can be used with the above recode options to define an INFO key name to keep in the output file. This option can be used multiple times to keep more of the INFO fields. The second option is used to keep all INFO values in the original file. |
Workflow Description Language¶
version development
import "tools/vardict_germline_1_6_0.wdl" as V
import "tools/bcftoolsAnnotate_v1_5.wdl" as B
import "tools/bgzip_1_2_1.wdl" as B2
import "tools/tabix_1_2_1.wdl" as T
import "tools/SplitMultiAllele_v0_5772.wdl" as S
import "tools/trimIUPAC_0_0_5.wdl" as T2
import "tools/VcfTools_0_1_16.wdl" as V2
workflow vardictGermlineVariantCaller {
input {
File bam
File bam_bai
File intervals
String sample_name
Float? allele_freq_threshold = 0.5
File header_lines
File reference
File reference_fai
File reference_amb
File reference_ann
File reference_bwt
File reference_pac
File reference_sa
File reference_dict
Boolean? vardict_chromNamesAreNumbers = true
Boolean? vardict_vcfFormat = true
Int? vardict_chromColumn = 1
Int? vardict_regStartCol = 2
Int? vardict_geneEndCol = 3
Boolean? compressvcf_stdout = true
Boolean? filterpass_removeFileteredAll = true
Boolean? filterpass_recode = true
Boolean? filterpass_recodeINFOAll = true
}
call V.vardict_germline as vardict {
input:
intervals=intervals,
bam=bam,
bam_bai=bam_bai,
reference=reference,
reference_fai=reference_fai,
chromNamesAreNumbers=select_first([vardict_chromNamesAreNumbers, true]),
chromColumn=select_first([vardict_chromColumn, 1]),
geneEndCol=select_first([vardict_geneEndCol, 3]),
alleleFreqThreshold=select_first([allele_freq_threshold, 0.5]),
sampleName=sample_name,
regStartCol=select_first([vardict_regStartCol, 2]),
vcfFormat=select_first([vardict_vcfFormat, true]),
var2vcfSampleName=sample_name,
var2vcfAlleleFreqThreshold=select_first([allele_freq_threshold, 0.5])
}
call B.bcftoolsAnnotate as annotate {
input:
vcf=vardict.out,
headerLines=header_lines
}
call B2.bgzip as compressvcf {
input:
file=annotate.out,
stdout=select_first([compressvcf_stdout, true])
}
call T.tabix as tabixvcf {
input:
inp=compressvcf.out
}
call S.SplitMultiAllele as splitnormalisevcf {
input:
vcf=annotate.out,
reference=reference,
reference_fai=reference_fai,
reference_amb=reference_amb,
reference_ann=reference_ann,
reference_bwt=reference_bwt,
reference_pac=reference_pac,
reference_sa=reference_sa,
reference_dict=reference_dict
}
call T2.trimIUPAC as trim {
input:
vcf=splitnormalisevcf.out
}
call V2.VcfTools as filterpass {
input:
vcf=trim.out,
removeFileteredAll=select_first([filterpass_removeFileteredAll, true]),
recode=select_first([filterpass_recode, true]),
recodeINFOAll=select_first([filterpass_recodeINFOAll, true])
}
output {
File variants = tabixvcf.out
File variants_tbi = tabixvcf.out_tbi
File out = filterpass.out
}
}
Common Workflow Language¶
#!/usr/bin/env cwl-runner
class: Workflow
cwlVersion: v1.2
label: Vardict Germline Variant Caller
doc: ''
requirements:
- class: InlineJavascriptRequirement
- class: StepInputExpressionRequirement
inputs:
- id: bam
type: File
secondaryFiles:
- pattern: .bai
- id: intervals
type: File
- id: sample_name
type: string
- id: allele_freq_threshold
type: float
default: 0.5
- id: header_lines
type: File
- id: reference
type: File
secondaryFiles:
- pattern: .fai
- pattern: .amb
- pattern: .ann
- pattern: .bwt
- pattern: .pac
- pattern: .sa
- pattern: ^.dict
- id: vardict_chromNamesAreNumbers
doc: Indicate the chromosome names are just numbers, such as 1, 2, not chr1, chr2
type: boolean
default: true
- id: vardict_vcfFormat
doc: VCF format output
type: boolean
default: true
- id: vardict_chromColumn
doc: The column for chromosome
type: int
default: 1
- id: vardict_regStartCol
doc: The column for region start, e.g. gene start
type: int
default: 2
- id: vardict_geneEndCol
doc: The column for region end, e.g. gene end
type: int
default: 3
- id: compressvcf_stdout
doc: 'c: Write to standard output, keep original files unchanged.'
type: boolean
default: true
- id: filterpass_removeFileteredAll
doc: Removes all sites with a FILTER flag other than PASS.
type: boolean
default: true
- id: filterpass_recode
doc: ''
type: boolean
default: true
- id: filterpass_recodeINFOAll
doc: |-
These options can be used with the above recode options to define an INFO key name to keep in the output file. This option can be used multiple times to keep more of the INFO fields. The second option is used to keep all INFO values in the original file.
type: boolean
default: true
outputs:
- id: variants
type: File
secondaryFiles:
- pattern: .tbi
outputSource: tabixvcf/out
- id: out
type: File
outputSource: filterpass/out
steps:
- id: vardict
label: VarDict (Germline)
in:
- id: intervals
source: intervals
- id: bam
source: bam
- id: reference
source: reference
- id: chromNamesAreNumbers
source: vardict_chromNamesAreNumbers
- id: chromColumn
source: vardict_chromColumn
- id: geneEndCol
source: vardict_geneEndCol
- id: alleleFreqThreshold
source: allele_freq_threshold
- id: sampleName
source: sample_name
- id: regStartCol
source: vardict_regStartCol
- id: vcfFormat
source: vardict_vcfFormat
- id: var2vcfSampleName
source: sample_name
- id: var2vcfAlleleFreqThreshold
source: allele_freq_threshold
run: tools/vardict_germline_1_6_0.cwl
out:
- id: out
- id: annotate
label: 'BCFTools: Annotate'
in:
- id: vcf
source: vardict/out
- id: headerLines
source: header_lines
run: tools/bcftoolsAnnotate_v1_5.cwl
out:
- id: out
- id: compressvcf
label: BGZip
in:
- id: file
source: annotate/out
- id: stdout
source: compressvcf_stdout
run: tools/bgzip_1_2_1.cwl
out:
- id: out
- id: tabixvcf
label: Tabix
in:
- id: inp
source: compressvcf/out
run: tools/tabix_1_2_1.cwl
out:
- id: out
- id: splitnormalisevcf
label: Split Multiple Alleles
in:
- id: vcf
source: annotate/out
- id: reference
source: reference
run: tools/SplitMultiAllele_v0_5772.cwl
out:
- id: out
- id: trim
label: Trim IUPAC Bases
in:
- id: vcf
source: splitnormalisevcf/out
run: tools/trimIUPAC_0_0_5.cwl
out:
- id: out
- id: filterpass
label: VcfTools
in:
- id: vcf
source: trim/out
- id: removeFileteredAll
source: filterpass_removeFileteredAll
- id: recode
source: filterpass_recode
- id: recodeINFOAll
source: filterpass_recodeINFOAll
run: tools/VcfTools_0_1_16.cwl
out:
- id: out
id: vardictGermlineVariantCaller